Глава III.6.
Динамика атомного остова и электронная структура эндо- и экзоэдральных
комплексов фуллеренов с легкими элементами
В
настоящее время известно уже большое количество соединений фуллерена с
металлами (см., например [1-3]). Все это многообразие делится на два больших
класса - эндоэдральные комплексы, когда атомы металла находятся внутри
фуллереновых полиэдров, и экзоэдральные - когда атомы металла находятся
снаружи. Оба класса привлекают к себе пристальное внимание исследователей
своими уникальными химическими и физическими свойствами, в частности -
магнитными и сверхпроводящими. В настоящее время пожалуй самыми интересными металло-фуллереновыми
объектами являются экзо- и эндоэдральные комплексы фуллеренов со щелочными
металлами. Это объясняется, в частности тем, что соединения типа K3C60
и Rb3C60 [1, 4] являются сверхпроводниками с достаточно
высокими температурами сверхпроводящего перехода, доходящими до 55К. В
последнее время был также разработан и элегантный способ синтеза эндоэдралов
Li@C60, Li2@C60 и Li3@C60,
заключающийся в том, что фуллерит C60 облучается пучком ионов лития
с энергией до 30 эВ [5].
Комплексы
фуллеренов с металлами интенсивно и уже достаточно долго изучаются как
экспериментальными (см., например, [5-12]), так и теоретическими (см., например
[1, 3, 13-27]) методами. Методами электронной спектроскопии исследовалась
электронная структура металлокомплексов. Так, в [9] исследовались
фотоэлектронные спектры и спектры обращенной фотоэмиссии экзоэдральных
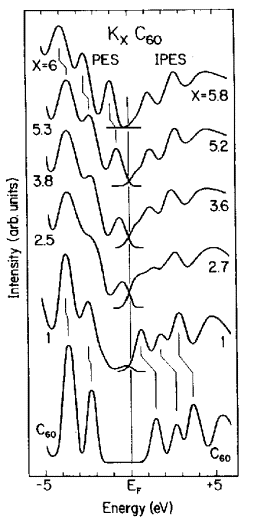
соединений типа KxC60 (Рис. 6.1 [9]). Как видно,
заполнение нижней вакантной орбитали С60 при допировании щелочным
металлом (в данном случае - калием) приводит не только к смещению занятых и
вакантных состояний, но и к существенному изменению формы полос и сложному
поведению уровня Ферми. Переход от чистого C60 к KC60
приводит к исчезновению запрещенной щели за счет возникновения внедренного
состояния с одной стороны и существенного (порядка 1 эВ) смещения первой
особенности спектра обращенной фотоэмиссии - с другой. Дальнейшее повышение
степени допирования приводит к монотонному увеличению плотности внедренного
электронного состояния, тогда как вакантное состояние ведет себя более сложно -
при степени допирования 2.5 оно максимально смещается вниз по энергии (на 1.5
эВ по сравнению с C60) и затем опять начинает двигаться вверх по
энергетической шкале и при x»6 вновь появляется запрещенная щель.
|
|
Рисунок 6.1. Фотоэлектронные
(PES) спектры и спектры обращенной
фотоэмиссии (IPES) [9] соединений типа KxC60. При x=1 спектральная интенсивность
фотоэлектронного спектра на уровне ферми мала, а первая особенность спектра
обращенной фотоэмиссии большая, однако с ростом степени допирования
интенсивность фотоэлектронного спектра возрастает, а первой особенности
спектра обращенной фотоэмиссии - падает. |
Сейчас
известно, что полуэмпирические квантово-химические методы хорошо описывают электронную
структуру и равновесную атомную геометрию как самой молекулы С60,
так и ее производных, допированных щелочными металлами. Так, в [28]
сравнивались результаты, полученные методом Хартри-Фока в базисе 3-21G с
экспериментальными фотоэлектронными спектрами и результатами полуэмпирических
расчетов методами PM3 и MNDO для эндоэдральных комплексов С60 с
ионами лития. Было показано, что полуэмпирические методы хорошо повторяют
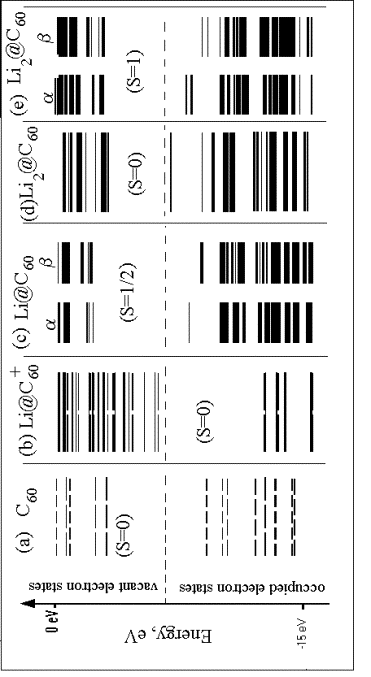
результаты неэмпирических расчетов электронной структуры (Рис. 6.2) и адекватно
описывают экспериментальные фотоэлектронные данные. Связь ионов с углеродной
стенкой носит ионный характер (заряд иона щелочного металла порядка +0.6), так
как углеродный полиэдр относительно щелочного металла является окислителем.
Верхней заполненной орбиталью (ВЗМО) в молекуле С60 является
пятикратно вырожденнй уровень (Рис. 6.2а). Размер иона лития существенно меньше
внутренней полости молекулы С60, поэтому он координируется к стенке
полиэдра. Это снижает симметрию объекта и снимает вырождение с ВЗМО, расщепляя
ее на уровни с более низким вырождением (Рис. 6.2b – 2e), с закрытой (случаи, в
частности, комплексов ![]() (Рис. 2b) и
(Рис. 2b) и ![]() (Рис. 6.2d)) и
открытой (в частности,
(Рис. 6.2d)) и
открытой (в частности, ![]() (полный спин
(полный спин ![]() , Рис. 8.2c) и
, Рис. 8.2c) и ![]() (полный спин
(полный спин ![]() (Рис. 6.2e))
оболочками с мультиплетностью по спину (2S+1) соответственно 1, 2 и 3. Для
систем с тремя добавочными электронами (комплекс
(Рис. 6.2e))
оболочками с мультиплетностью по спину (2S+1) соответственно 1, 2 и 3. Для
систем с тремя добавочными электронами (комплекс ![]() ) мультиплетность может принимать значение 2 или 4.
) мультиплетность может принимать значение 2 или 4.
|
|
Рисунок 6.2. Энергетическая диаграмма электронных уровней
комплексов |
Впрочем,
такой результат не вызывает удивления, особенно в случае эндоэдральных
комплексов. Дело в том, что все полуэмпирические методы хорошо описывают
системы на основе углерода с сильными химическими связями, какими являются
фуллерены и их эндо-производные с щелочными металлами. Из-за своей уникальной
электронной структуры полиэдр фуллерена является окислителем для щелочных
металлов и «перетягивает» к себе s-электроны
металла. Таким образом, связь внутри молекулы становится в основном ионной
(заряд иона щелочного металла порядка +0.6). В случае же, когда внутри
углеродного полиэдра помещаются более одного атома-гостя (исключая молекулу Н2),
система вдобавок ко всему становится напряженной, так как внутреннего объема уже
не хватает на их свободное размещение.
Другим
информативным методом исследования электронной и атомной структур как чистых
соединений, так и их комплексов с металлами является спектроскопия ЯМР и ЭПР.
Так, практически сразу же после синтеза молекулы С60 при помощи
спектроскопии ЯМР было установлено, что в твердом теле при комнатной
температуре молекулы С60 вращаются [29-31] с частотой порядка 1011
сек-1, при этом оси вращения молекул направлены случайно. При
понижении температуры до 250К происходит замедление и упорядочение вращения,
которое теперь уже происходит только вдоль одной оси [32, 33].
Аналогичные
результаты по вращению молекул C60 вдоль одной выделенной оси
кристаллов K3C60 и Rb3C60 были
получены в работе [6] при помощи спектроскопии ЯМР. В этой работе также
указывалось, что замораживание вращения при определенной температуре играет
важную роль в фазовых переходах подобных веществ.
Необходимо
отметить другую важную работу в этой области [11]. В ней молекулярная и
межмолекулярная динамика исследовалась для твердого тела CeLa@C80.
Это соединение получалось в процессе b-распада одного из атомов 140La, помещенных внутрь углеродного
полиэдра. В этой работе были зафиксированы не только вращательные движения
самих молекул, которые скачкообразно замораживались при 160К, но и вращательные
движение атомов Се, которые продолжались до температур порядка 40К.
Быстрая
миграция иона La внутри фуллерена была изучена и в методе молекулярной динамики
на основе потенциала, полученного в LDA-подходе [34]. Было показано, что La
очень быстро тангенциально двигается относительно углеродного полиэдра и
совершает один оборот примерно за одну пикосекунду. Аналогичные результаты были
получены авторами [34, 35] и для эндоэдрала La@C82. Эти результаты позволили
сделать вывод, что ряд экспериментов по электронной и атомной структуре
необходимо интерпретировать по другому, с учетом вращения. Так, в частности,
спектроскопией ЭПР становится невозможно извлекать необходимую информацию, так
как характерные времена для ЭПР-процессов на порядок больше, нежели чем период
вращения иона лантана внутри полиэдра.
Очевидно,
что не все атомы и молекулы-гости могут двигаться внутри углеродных полиэдров.
Несмотря на то, что на эти экзотические соединения было обращено пристальное
внимание как экспериментаторов, так и теоретиков, в частности, вопрос - о
способе координации атомов и молекул-гостей, которые остаются неподвижными
внутри полиэдра, в общем виде так и не был решен. Действительно, до настоящего
времени были получены лишь единичные экспериментальные сведения о структуре
твердых тел на основе эндоэдралов, как например структура Y@C82, для
которого известно [10], что атом иттрия жестко координирован изнутри к
углеродной стенке, а сами эндоэдральные молекулы в молекулярном твердом теле
координированы способом «голова к хвосту». Эти данные были получены на
синхротронном источнике излучения методами рентгеновской порошковой дифракции и
максимальной энтропии (MEM). Как было показано, атом иттрия удален от центра
полиэдра С82 на расстояние 3.14 ангстрем. Также было показано, что
вращение эндоэдрального комплекса в кристаллической решетке подавлено, тогда
как С82 вращается свободно. Расстояние самого атома-гостя до
углеродной стенки составляет при этом около 2.9 ангстрем, что немного больше,
чем ранее предсказывалось квантово-химическими методами.
По
вполне понятным причинам, в теоретическом плане способы координации атомов -
гостей изучены в настоящее время гораздо лучше, нежели чем в экспериментальном.
Было показано, что некоторые атомы и ионы, к примеру He, K+ [16, 17,
25], должны координироваться в центре углеродного полиэдра, а некоторые другие,
такие, как Li+, Na+ - около углеродной стенки [13, 16,
36]. Из самых общих положений ясно, что для одного атома-гостя существует 5
способов координации (к центрам шестиугольника, пятиугольника, ребра
шестиугольник-шестиугольник, ребра пятиугольник-шестиугольник и к вершине
усеченного икосаэдра). Очевидно, что при наличии более одного атома-гостя
способов координации внутри углеродного полиэдра может быть еще больше.
Необходимо отметить интересную с теоретической точки зрения работу [14], в которой было показано, что атомы-гости могут туннелировать из одного в другой минимум многоямной потенциальной поверхности внутренней углеродной стенки. Однако эта задача была решена в общем виде с использованием модельного гамильтониана в предположении, что потенциальная поверхность движения атома-гостя имеет двадцать минимумов, каждый из которых расположен над центром одного из шестиугольников. При этом природа химического взаимодействия углеродной стенки с атомом-гостем не рассматривалась.
В
этой главе электронная и атомные структуры и динамические свойства ряда
комплексов фуллерена с атомами лития, гелия и молекулой водорода исследовались
стандартными полуэмпирическими квантово-химическими методами PM3, MNDO и INDO,
неэмпирическим методом Хартри-Фока в базисе 3-21G и методом молекулярной
динамики на основе соответствующих потенциалов. Для проведения расчетов
использовались программы GAMES [37] (электронная структура и равновесная
атомная геометрия) и демонстрационная версия программы HyperChem 5.02
(электронная структура, равновесная атомная геометрия и молекулярная динамика).
Такой широкий выбор квантово-химических методов обусловлен рядом причин:
1.
Необходимо убедится, что
потенциалы, получаемые в различных квантово-химических методах (как
неэмпирических, так и полуэмпирических), дают согласующиеся друг с другом
результаты в методе молекулярной динамики.
2.
К сожалению, ни один из
полуэмпирических методов не имеет параметризации для всех атомов, входящих в состав выбранных объектов.
3.
Проведение
молекулярно-динамических расчетов в квантово-химическом ab initio подходе даже в малом базисе типа 3-21G для больших
(порядка нескольких десятков атомов углерода, типичный пример – молекула С60)
систем в настоящее время на доступных компьютерах невозможно.
Как было показано выше, выбор полуэмпирических квантово-химических подходов для массированного молекулярно-динамического моделирования поведения подобных систем представляется вполне оправданным и адекватным. Во всех случаях рассчитанная равновесная геометрия использовалась в качестве начальной для молекулярно-динамического расчета, который проводился в приближении, что исследуемый объект находится в вакууме.
В главе рассматриваются следующие объекты:
1/. Молекула C60 икосаэдрической симметрии.
Электронная структура и равновесная геометрия рассчитывались в приближении
ограниченного метода Хартри-Фока неэмпирическим методом Хартри-Фока в базисе
3-21G (C60_3-21G.hin) и полуэмпирическим методом PM3 (C60.hin). Длина связи 6-6 в ab
initio подходе и в методе РМ3 составила 0.1367 и 0.1384 nm соответственно, а длина связи 6-5 –
0.1453 и 0.1457 nm. Молекулярная
динамика рассчитывалась в потенциале полуэмпирического метода PM3 без учета
симметрии (C60_300K.hin).
2/. Молекула C36, симметрия D6h.
Электронная структура и равновесная геометрия рассчитывались в приближении
ограниченного метода Хартри-Фока неэмпирическим методом Хартри-Фока в базисе
3-21G (c36_3-21G.hin) и полуэмпирическим методом PM3 (директория Databank of structures/Fullerens/C36,
файлы, содержащие результаты расчетов различных изомеров (D3h, D6h,
D2d, C2v симметрии) молекулы С36). Длина
связей в ab initio подходе и в методе
РМ3 (симметрия D6h) составили: 1-й тип (6-6) 0.1.393 и 0.1411 nm соответственно, 2-й тип (6-5) 0.1438
и 0.1437 nm, 3-й тип (6-5) 0.1415 и
0.1430 nm, 4-й тип (5-5) 0.1485 и
0.1499 nm. Для проверки этих
результатов были проведены расчеты в базисе 6-31G* и методом DFT
B3LYP. Оба этих расчета подтвердили корректность расчетов в базисе 3-21G и
полуэмпирическим методом PM3. Молекулярная динамика рассчитывалась в
потенциалах полуэмпирического метода PM3 (c36_d6h_300K.hin) без учета симметрии.
3/. Эндоэдральный комплекс Li2@C60
в различных зарядовых (отрицательный ион, нейтральный комплекс и положительно
заряженный ион) и спиновых состояниях (синглет, дублет, триплет). Электронная
структура и равновесная геометрия рассчитывались в приближении ограниченного и
неограниченного методов Хартри-Фока (в зависимости от зарядового и спинового
состояний) полуэмпирическим методом MNDO (более детальное изучение электронной
и атомной структур этого комплекса, в том числе – и неэмпирическим методом
Хартри-Фока, приведено в Главах 5, 6). Молекулярная динамика рассчитывалась в
потенциале полуэмпирического метода MNDO (Li2C60_C_C_78K.hin, Li2C60_C_C_79K.hin, Li2C60_C_C_300K.hin) (нейтральный комплекс, синглетное основное состояние), (Li2@C60_+_300K.hin) (положительно заряженный ион, дублетное спиновое
состояние), (Li2@C60_-_300K.hin) (отрицательно заряженный ион, дублетное спиновое
состояние), (Li2@C60_M3_300K.hin) (нейтральный комплекс, триплетное состояние) без учета
симметрии.
4/. Экзоэдральный комплекс Li2C60,
(Набор файлов в директории Databank/Isomers _ Li) электронная структура и равновесная геометрия
рассчитывались в приближении ограниченного метода Хартри-Фока полуэмпирическим
методом MNDO (см. Главы 5, 6). Молекулярная динамика рассчитывалась в
потенциале полуэмпирического метода MNDO (Exo_Li2C60_300K) без учета симметрии.
5/. Эндо-экзоэдральный комплекс Li[Li@C60] (один из ионов
находится внутри полиэдра, а другой - снаружи), электронная структура и
равновесная геометрия рассчитывались в приближении ограниченного метода
Хартри-Фока полуэмпирическим методом MNDO, молекулярная динамика рассчитывалась
в потенциале полуэмпирического метода MNDO (Endo_Exo_Li2C60_300K) без учета симметрии.
6/. Эндоэдральный комплекс Li@C60 в различных зарядовых (отрицательный ион, положительный
ион и нейтральный комплекс) и спиновых (синглетное, дублетное и триплетное)
состояниях. Электронная структура и равновесная геометрия рассчитывались в
приближения ограниченного и неограниченного метода Хартри-Фока (соответственно
спиновому состоянию) полуэмпирическим методом MNDO (см. Главы 5, 6, Li+C60.hin). Молекулярная динамика рассчитывалась в потенциале
полуэмпирического метода MNDO (Li+C60_300K.hin) (положительно заряженный ион), (Li@C60_-_M1_300K.hin) и (Li@C60_-_M3_300K.hin) (отрицательно заряженный ион, синглетное (М1) и
триплетное М3) состояния), (C60Li_300K.hin) (нейтральный кластер, дублет)).
7/. Эндоэдральный комплекс Li3@C60 в различных зарядовых (отрицательный ион, положительный
ион и нейтральный комплекс) и спиновых (синглетное, дублетное, триплетное и квартетное)
состояниях. Электронная структура и равновесная геометрия рассчитывались в
приближения ограниченного и неограниченного метода Хартри-Фока (соответственно
спиновому состоянию) полуэмпирическим методом MNDO. Молекулярная динамика
рассчитывалась в потенциале полуэмпирического метода MNDO (Li3@C60_+_M1_300K_2ps.hin) и (Li3@C60_+_M3_300K.hin) (положительно заряженный ион, синглет (М1) и триплет
(М3) соответственно), (Li3@C60_-_M1_300K.hin) и (Li3@C60_-_M3_300K.hin) (отрицательно заряженный ион, синглетное (М1) и
триплетное (М3) состояния), (Li3C60_300K.hin) и (Li3@C60_M4_300K.hin) (нейтральный кластер, дублет и квартет (М4)).
8/. Низший фуллерен С26 и эндоэдральный комплекс H2@C26,
электронная структура и равновесная геометрия рассчитывались в приближении
ограниченного метода Хартри-Фока неэмпирическим методом Хартри-Фока в базисе
3-21G (c26_3-21G.hin) и (H2@c26_3-21G.hin). Структура полиэдра С26 сохранилась в
комплексе практически неизменной. Молекулярная динамика рассчитывалась в
потенциалах полуэмпирического метода PM3 (H2c26_300K.hin) и неэмпирическом потенциале 3-21G (H2c26_3-21G_300K.hin).
9/. Эндоэдральный
комплекс H2@C36, электронная структура и равновесная
геометрия рассчитывались в приближении ограниченного метода Хартри-Фока
неэмпирическим методом Хартри-Фока в базисе 3-21G (c36h2_3-21G.hin) и полуэмпирическим методом PM3 (H2c36_d6h.hin). Структура полиэдра С36 сохранилась в
комплексе практически неизменной. Молекулярная динамика рассчитывалась в
потенциале полуэмпирического метода PM3 (H2c36_d6h_300K.hin).
10/. Эндоэдральный
комплекс H2@C40, электронная структура и равновесная
геометрия рассчитывались в приближении ограниченного метода Хартри-Фока
полуэмпирическим методом PM3 (H2_C40_Td.hin). Структура полиэдра С40 выбиралась Td
симметрии. Молекулярная динамика рассчитывалась в потенциале полуэмпирического
метода PM3 (H2_C40_Td_300K.hin).
11/. Эндоэдральный
комплекс H2@C50, электронная структура и равновесная
геометрия рассчитывались в приближении ограниченного метода Хартри-Фока
полуэмпирическим методом PM3 PM3 (H2_C50_D5h.hin). Структура полиэдра С50 выбиралась D5h
симметрии. Длинны связей составили: 1-й тип (6-6) 0.1405 nm, 2-й тип (6-6) 0.1378 nm,
3-й тип (6-5) 0.1419 nm, 4-й тип
(6-5) 0.1474 nm, 5-й тип (6-5) 0.1467
nm, 6-й тип (5-5) 0.1481 nm. Молекулярная динамика рассчитывалась
в потенциале полуэмпирического метода PM3 (H2_C50_D5h_v2_300K.hin).
12/. Эндоэдральный
комплекс H2@C60, электронная структура и равновесная
геометрия рассчитывались в приближении ограниченного метода Хартри-Фока
полуэмпирическим методом PM3 PM3 (H2_C60.hin), молекулярная динамика рассчитывалась в потенциалах
полуэмпирического метода PM3 для температур 300К и 4К (H2_C60_300K.hin) и (H2_C60_4K.hin).
13/. Эндоэдральный
комплекс (H2)16@C60, электронная структура и
равновесная геометрия рассчитывались в приближении ограниченного метода
Хартри-Фока полуэмпирическим методом PM3 PM3 ([email protected]), молекулярная динамика рассчитывалась в потенциалах
полуэмпирического метода PM3 для температур 300К и 4К (H2_16@C60_300K.hin) и (H2_16@C60_4K.hin).
14/. Ряд эндоэдральных
комплексов Hen@C60, n=2,3,4 и отрицательно заряженного
комплекса He2@C60-. Электронная структура и
равновесная геометрия рассчитывались в приближении ограниченного метода
Хартри-Фока полуэмпирическим методом INDO (He2_C60.hin, He3_C60.hin, He4_C60.hin, He5_C60.hin, He6_C60.hin, He2@C60(-1).hin). Длина связи 6-6 составила 0.1397 nm, а длинна связи 6-5 0.1449 nm.
Молекулярная динамика рассчитывалась в потенциале полуэмпирического метода INDO
(He2_C60_300K.hin, He2_C60_4K.hin, He3_C60_4K.hin, He3_C60_6K.hin,
He4_C60_4K.hin,
He5_C60_4K.hin,
He6_C60_4K.hin,
He2@C60_-1_300K.hin) для температур 300, 6 и 4К соответственно.
Все расчеты комплексов с легкими элементами проводились без учета симметрии системы. Оптимизация геометрии этих комплексов проводилась во всех методах с параметром сходимости 0.01 ккал/моль/атом. Метод молекулярной динамики был описан в главе II этой книги.
Результаты молекулярно-динамических расчетов вышеперечисленных систем (исключая экзоэдральные комплексы) приведены в Табл. (1). Как видно, в том случае, когда рассчитываемая система обладает нулевым спином, атомы-гости под действием температуры начинают перемещаться из одного минимума многоямной потенциальной внутренней стенки углеродного полиэдра поверхности в другой минимум, приобретая при этом ненулевой орбитальный момент. Вращаются также и молекулы фуллеренов как целое.
Таблица 1
Тип структурной координации некоторых эндоэдральных комплексов
|
Комплекс |
Метод |
Заряд |
Мультиплетность по спину (2S+1) |
Полная энергия (ккал/моль) |
Тип поведения атомов-гостей |
|
|
RHF MNDO |
+1 |
1 |
-9238.6899 |
Наблюдается орбитальное движение |
|
|
UHF MNDO |
0 |
2 |
-9385.7744 |
Система жесткая |
|
|
RHF MNDO |
-1 |
1 |
-9461.3477 |
Наблюдается орбитальное движение |
|
|
UHF MNDO |
-1 |
3 |
-9459.1534 |
Система жесткая |
|
|
UHF MNDO |
+1 |
2 |
-9224.5732 |
Система жесткая |
|
|
RHF MNDO |
0 |
1 |
-9376.9805 |
Наблюдается орбитальное движение |
|
|
UHF MNDO |
0 |
3 |
-9376.0400 |
Система жесткая |
|
|
UHF MNDO |
-1 |
2 |
-9455.6463 |
Система жесткая |
|
|
RHF MNDO |
+1 |
1 |
-9162.7617 |
Наблюдается орбитальное движение |
|
|
UHF MNDO |
+1 |
3 |
-9212.5811 |
Система нежесткая, орбитального движения нет |
|
|
UHF MNDO |
0 |
2 |
-9352.0771 |
Система нежесткая, орбитального движения нет |
|
|
UHF MNDO |
0 |
4 |
-9361.2725 |
Система нежесткая, орбитального движения нет |
|
|
RHF MNDO |
-1 |
1 |
-9438.7076 |
Наблюдается орбитальное движение |
|
|
UHF MNDO |
-1 |
3 |
-9439.8832 |
Система нежесткая, орбитального движения нет |
|
|
RHF INDO |
0 |
1 |
-35263.3028 |
Наблюдается орбитальное движение |
|
|
UHF INDO |
-1 |
2 |
-35241.0413 |
Наблюдается орбитальное движение |
|
C60 |
PM3 |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
C36 |
PM3 |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(H2@C26) |
PM3, ab initio |
0 |
0 |
|
Наблюдается орбитальное движение сферы в обоих методах.
В ab
initio методе молекула-гость обладает ярко выраженым орбитальным
движением. |
|
(H2@C36) |
PM3, |
0 |
0 |
|
Наблюдается орбитальное движение сферы. В симметрии D3h
наблюдается орбитальное движение молекулы-гостя. |
|
(H2@C40) |
PM3 |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(H2@C50) |
PM3 |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(H2@C60) |
PM3 |
0 |
0 |
|
Наблюдается орбитальное движение сферы, поведение
молекулы-гостя более сложно |
|
(He3@C60) |
INDO |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(He4@C60) |
INDO |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(He5@C60) |
INDO |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
|
(He6@C60) |
INDO |
0 |
0 |
|
Наблюдается орбитальное движение сферы |
Частоты вращения некоторых комплексов приведены в Таблице 2.
Таблица 2
Частоты вращения
димера Li2 фуллерена С60 в эндоэдральных комплексах Li2@C60
и (Li2@C60)2.
|
Соединение, температура |
Частота вращения димера Li2, сек-1 |
Частота вращения фуллерена С60, сек-1 |
|
Li2@C60 при Т=79 К |
1*1012 |
2.5*109 |
|
Li2@C60 при Т=300 К |
2.5*1012 |
3.4*109 |
|
С60 при Т=300 К |
- |
7.9*109 |
|
Экспериментальная частота вращения газообразной молекулы С60
при 300 К [29-31] |
- |
3.3*1011 |
|
Экспериментальная частота вращения молекулы С60 в фуллерите при 300К [29-31] |
- |
1.1*1011 |
|
Экспериментальная частота вращения молекулы С60 в фуллерите ниже 260 К [29-31] |
- |
5.0*108 |
Электронная
структура молекулы С60 в настоящее время хорошо известна (см.,
например, главу IV этой книги), известны также и ее динамические свойства в
различных условиях и состояниях (см. Табл. 1, 2). Как показали наши
молекулярно-динамические расчеты, наблюдается вращательное движение молекулы С60
вокруг своего центра масс как единого целого. Период вращения ![]() и частота
и частота ![]() определялись по
изменению координат атомов углеродного полиэдра во времени. Расчеты свободной
молекулы С60 показали, что частота ее вращения при 300 К составляет
0.79*1010 сек-1. Экспериментальные частоты, определенные
по спектрам ЯМР для газообразной молекулы С60 при 300 К составляет -
3.3*1011 сек-1 [29-30], для молекулы С60 при
300 К в твердом теле - 1.1*1011 сек-1, а для С60
ниже 260 К в твердом теле - 5.0*108 сек-1. Как видно,
наши теоретические расчеты методом неэмпирической молекулярной динамики
качественно описывают эффект вращения молекул С60. Отличие в частотах
вращения, очевидно, вызваны тем фактом, что полуэмпирические методы расчета,
как правило, завышают энергию и константу связи и, тем самым, недооценивают
изменение межъядерных расстояний при увеличении температуры.
определялись по
изменению координат атомов углеродного полиэдра во времени. Расчеты свободной
молекулы С60 показали, что частота ее вращения при 300 К составляет
0.79*1010 сек-1. Экспериментальные частоты, определенные
по спектрам ЯМР для газообразной молекулы С60 при 300 К составляет -
3.3*1011 сек-1 [29-30], для молекулы С60 при
300 К в твердом теле - 1.1*1011 сек-1, а для С60
ниже 260 К в твердом теле - 5.0*108 сек-1. Как видно,
наши теоретические расчеты методом неэмпирической молекулярной динамики
качественно описывают эффект вращения молекул С60. Отличие в частотах
вращения, очевидно, вызваны тем фактом, что полуэмпирические методы расчета,
как правило, завышают энергию и константу связи и, тем самым, недооценивают
изменение межъядерных расстояний при увеличении температуры.
Вращение
и возникновение ненулевого орбитального момента всей молекулы при повышении
температуры объясняется изменением полного электронного орбитального момента.
Действительно, полный момент системы (электронный плюс ядерный) должен
сохраняться, однако при повышении температуры изменяются эффективные
межъядерные расстояния, следовательно в общем виде можно записать:
![]() .
.
Это
неравенство имеет место, поскольку ![]() , так как при изменении температуры изменяется набор
координат ядер (
, так как при изменении температуры изменяется набор
координат ядер (![]() ), которые входят в полноэлектронную волновую функцию в виде
параметров. Следовательно, вся система должна компенсировать изменение
электронного орбитального момента изменением ионного орбитального момента, что
и проявляется во вращении молекул как целого. Однако в том случае, если полный
спин системы не равен нулю (исключая эндоэдральный комплекс с димером Не2,
о котором речь пойдет ниже), поведение атомов-гостей приобретает принципиально
другой характер (см. Табл. 1). В этом случае атомы-гости становятся или жестко
координированы, или же хаотично перемещаются из одного минимума потенциальной
поверхности в другой, при этом не проявляя орбитального движения. Причем это
свойство не зависит, находится ли система в основном состоянии, или же в
возбужденном (Табл. 1). Таким образом, единственная причина, которая фиксирует
атомную геометрию, является требование сохранения полного момента (орбитального
+ спинового) системы. Действительно, для эндоэдральных комплексов, содержащих
один или два иона лития, естественной выделенной осью проекции электронного
спина является ось, проходящая через центр полиэдра и один/два иона лития.
Орбитальное движение в этом случае должно приводить к изменению ориентации этой
оси в пространстве и, в конечном итоге, если спин системы не равен нулю, к
изменению его направления. Для эндоэдралов, содержащих три иона лития
выделенной осью является ось, проходящая через центр треугольника Li3.
Нужно помнить, что данная система является сильно напряженной, а за счет того,
что положительные ионы лития находятся достаточно близко, анизотропия системы
частично снимается и становится возможным перескоки ионов из одной
потенциальной ямы в другую. Однако, и в этом случае, несмотря на то, что
атомная система становится нежесткой, тем не менее она не приобретает
орбитального движения, а перескоки ионов по минимумам потенцияльной поверхности
происходят хаотично и около начального положения, задаваемого в расчетах.
Единственным исключением из всех вышеприведенных случаев является эндоэдральный
комплекс с димером гелия. Не смотря на то, что анион
), которые входят в полноэлектронную волновую функцию в виде
параметров. Следовательно, вся система должна компенсировать изменение
электронного орбитального момента изменением ионного орбитального момента, что
и проявляется во вращении молекул как целого. Однако в том случае, если полный
спин системы не равен нулю (исключая эндоэдральный комплекс с димером Не2,
о котором речь пойдет ниже), поведение атомов-гостей приобретает принципиально
другой характер (см. Табл. 1). В этом случае атомы-гости становятся или жестко
координированы, или же хаотично перемещаются из одного минимума потенциальной
поверхности в другой, при этом не проявляя орбитального движения. Причем это
свойство не зависит, находится ли система в основном состоянии, или же в
возбужденном (Табл. 1). Таким образом, единственная причина, которая фиксирует
атомную геометрию, является требование сохранения полного момента (орбитального
+ спинового) системы. Действительно, для эндоэдральных комплексов, содержащих
один или два иона лития, естественной выделенной осью проекции электронного
спина является ось, проходящая через центр полиэдра и один/два иона лития.
Орбитальное движение в этом случае должно приводить к изменению ориентации этой
оси в пространстве и, в конечном итоге, если спин системы не равен нулю, к
изменению его направления. Для эндоэдралов, содержащих три иона лития
выделенной осью является ось, проходящая через центр треугольника Li3.
Нужно помнить, что данная система является сильно напряженной, а за счет того,
что положительные ионы лития находятся достаточно близко, анизотропия системы
частично снимается и становится возможным перескоки ионов из одной
потенциальной ямы в другую. Однако, и в этом случае, несмотря на то, что
атомная система становится нежесткой, тем не менее она не приобретает
орбитального движения, а перескоки ионов по минимумам потенцияльной поверхности
происходят хаотично и около начального положения, задаваемого в расчетах.
Единственным исключением из всех вышеприведенных случаев является эндоэдральный
комплекс с димером гелия. Не смотря на то, что анион ![]() обладает ненулевым
электронным спином, тем не менее, димер гелия при ненулевой температуре
характеризуется орбитальным движением внутри углеродного полиэдра. Это связано
с тем, что атомы гелия химически не связаны ни с углеродной стенкой, ни с друг
другом и, следовательно, их движение не пораждает волны электронной плотности
на поверхности углеродного полиэдра и, следовательно, не наблюдается изменения
ориентации электронного спина.
обладает ненулевым
электронным спином, тем не менее, димер гелия при ненулевой температуре
характеризуется орбитальным движением внутри углеродного полиэдра. Это связано
с тем, что атомы гелия химически не связаны ни с углеродной стенкой, ни с друг
другом и, следовательно, их движение не пораждает волны электронной плотности
на поверхности углеродного полиэдра и, следовательно, не наблюдается изменения
ориентации электронного спина.
Неэмпирические
расчеты низших фуллеренов C26 и С36 в базисе 3-21G при
температуре 300К показали, что этот эффект воспроизводится и в том случае,
когда в методе молекулярной динамики используется потенциал, полученный ab initio методом. Из-за того, что
такого типа расчеты требуют очень большого машинного ресурса, оценить частоту
вращения молекулы не удалось. Дело в том, что из-за особенностей алгоритма
метода молекулярной динамики с использованием системы термостатов система как
бы «разогревается» во времени. Типичное время разогрева составляет порядка 0.2
– 0.3 ps, а для корректной оценки
периода вращения необходимо проводить расчет длительностью 0.5 ps. В настоящее время мощности
современных стандартных компьютеров не позволяют проводить столь длительной
эмитации поведения системы. В нашем случае был проведен расчет длительностью
0.1 ps, что потребовало около двух
недель непрерывной работы компьютера P-II 450 Dual 256 Mb RAM. Расчет частоты
вращения С36 с использованием полуэмпирического потенциала,
полученного в рамках метода PM3, показал, что частота вращения несколько выше,
нежели чем для С60, и равна 3.2 *1010 сек-1.
Ab initio расчет в базисе 3-21G равновесной атомной структуры
гипотетического эндоэдральных комплексов H2@C26 и H2@C36
показали, что молекула водорода располагается в центре углеродного полиэдра.
Расчеты методом молекулярной динамики при температуре 300К также
продемонстрировали нежесткость координации молекулы Н2 внутри
полиэдра. Молекула водорода свободновращеется даже внутри такой небольшой структуры,
каковой является молекула С26.
Расчет
динамики гипотетических систем H2@C40 H2@C50
показал, что частота вращения
молекулы водорода в этих эндоэдральных комплексах несколько снижается - до 3*1013
сек-1, что объясняется наличием плато на потенциальной поверхности
внутри углеродного полиэдра. Молекула водорода во время движения смещается на
0.09 нм. от центра вдоль длинной оси C50.
Расчет
динамики эндоэдрального комплекса H2@C60 при 300К
показал, что молекула водорода в нем не вращается, а хаотично двигается внутри
фуллерена за счет температурных флюктуаций, что объясняется наличием большого
плато на потенциальной поверхности внутри углеродного полиэдра во всех
направлениях, которое существенно превышает размеры молекулы водорода. Так,
смещения молекулы-гостя достигают 0.1 нм. от центра C60. Расчет
динамики системы при 4К показал, что амплитуда движений молекулы водорода
снижается до 0.01 нм., однако система все равно остается нежесткой.
Расчеты
молекулярной динамики других интересных эндоэдральных комплексов фуллеренов с
атомами гелия (Hen@C60 , n=2, 3, 4) в полуэмпирическом
потенциале INDO показал, что подобные системы остаются нежесткими при Т=4К,
причем частоты вращения атомов гелия оцениваются для этих объектов в пределах 3¸5*1012 cек-1. Повышение температуры
до 300К приводит к заметному (до 6¸7*1012 cек-1) повышению частоты вращения гелиев.
Необходимо отметить, что внутри углеродного полиэдра фактически существуют
молекулы Hen, причем во всех случаях межъядерное расстояние равно
0.18 нм. (Нужно отметить, что расчет ван-дер-ваальсового димера He2
методом INDO дает межъядерное расстояние 0.50 нм., а в ab initio подходе (базис 6-31G) - 0.32 нм.).
Расчеты
электронной и атомной структур комплексов с литием показал, что ионы лития в
эндоэдральном комплексе Li2@C60 координируются к центрам
противоположных шестиугольников. Ионы лежат друг против друга таким образом,
что расстояние Li-Li - 0.299 nm, а расстояние Li-С (до углеродов,
принадлежащих шестиугольникам) - 0.328 nm.
Координация
литиев в экзоэдральном комплексе Li2C60 может идти как по
центру шестиугольника, так и по центру пятиугольника, причем расстояние от иона
лития до атомов углерода шестиугольника составляют 0.232 nm, а до атома углерода пятиугольника - 0.234 nm.
В
эндо-экзоэдральном комплексе Li[Li@C60] экзоэдральный ион
координировался к центру шестиугольника с расстоянием Li-С, равным 0.231 нм, а
эндоэдральный - к центру шестиугольника, соседствующего с шестиугольником, к
которому координируется внешнесферный литий, причем расстояние эндоэдральный
литий - атом углерода составляет 0.241 nm.
Координация
лития в комплексе Li@C60+ происходит по центру одного из
шестиугольников, причем расстояние Li-С равно 0.2405 nm, а расстояние ион лития - центр шестиугольника - 0.1909 nm.
Расчеты
методом молекулярной динамики в полуэмпирическом потенциале показывают, что при
температуре 4 К эндоэдральные ионы лития в комплексе Li2@C60
«примораживаются» к углеродной стенке. При температуре выше 79 К наблюдается
динамический переход, при котором ионы выходят из равновесной геометрии и
начинают вращаться внутри полиэдра с частотой 1.0*1012 сек-1,
при этом вращается и сама углеродная полиэдр с частотой 2.5*109 сек-1.
(Необходимо отметить, что эта температура динамического перехода является всего
лишь оценкой высоты потенциального барьера, а не термодинамической
характеристикой. Все расчеты, как это уже говорилось выше, проводились методом
молекулярной динамики, который не учитывает в полной мере температурные
флюктуации и фактически моделирует состояние термодинамического равновесия.)
При температуре 300 К частота вращения увеличивается и для литиев она
составляет 2.5*1012сек-1 и 3.4*109 сек-1
для углеродного полиэдра (см. Таблицу 2).
Как
показали аналогичные молекулярно-динамические расчеты, экзоэдральный комплекс
Li2C60 - жесткий, вплоть до температуры 300 К. Внешние
ионы лития только колеблются вблизи своих равновесных геометрий над центрами
как шестиугольника, так и пятиугольника.
Поведение
эндоэдрального иона в эндоэкзоэдральном комплексе Li[Li@C60] существенно более
сложно: при 77 К он меняет свою координацию с центра шестиугольника на ребро
шестиугольник-шестиугольник, принадлежащему обеим соседствующим
шестиугольникам, к которым при 4 К координировались экзо- и эндоэдральные ионы.
При 300 К эндоэдральный ион начинает мигрировать в телесном угле порядка 30° в области координации экзоэдрального лития.
Расчеты
методом молекулярной динамики в полуэмпирическом потенциале показывают, что при
температуре 300 К эндоэдральный ион лития в комплексе (Li@C60)+
движется с частотой порядка 5*1012 сек-1.
Проследим
теперь зависимость электронной структуры от динамических свойств эндоэдрального
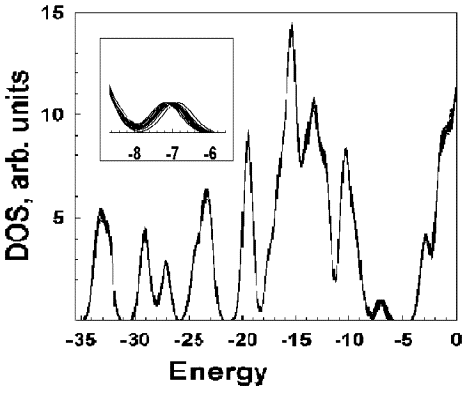
комплекса Li2@C60. На Рис. 8.3 представлены полные
плотности состояний, построенных для 16 стоп-кадров с интервалом 0.01 ps
динамического кино для этого комплекса, рассчитанного при 300К. Как видно,
происходит «заплывание» внедренного электронного состояния, образованного за
счет дополнительных электронов, поставляемых атомами лития. Изменение энергии
верхней заполненной орбитали, обусловленное изменением координации ионов лития
под действием температурных флюктуаций, достаточно велико - порядка 1 эВ.
Движение ионов лития вызывает и волну поляризации на углеродном полиэдре. Так,
вслед за движением ионов лития меняют свой заряд и атомы углерода,
малликеновские заряды которых могут меняться от нескольких сотых единицы
электронного заряда до пятнадцати сотых электронного заряда.
|
|
|
Рисунок 6.3. Электронная структура 16 стоп-кадров динамического
кино, наложенных друг на друга и сделанных с шагом 0.01 ps рассчитанного для
комплекса Li2@C60 при температуре 300К. Видно, что
верхняя заполненная орбиталь (внедренное электронное состояние) меняет свое
положение с амплитудой 1 эВ. На вставке изображено увеличенное внедреное
состояние. |
Таким
образом, расчеты методом неэмпирической молекулярной динамики показывают:
1/. Эндоэдральные комплексы фуллеренов с закрытыми оболочками
и легкими атомами и молекулами-гостями, не связанными ковалентно с углеродными
стенками, являются системами нежесткими.
2/. Это свойство объясняется невысокими (порядка десятков К)
потенциальными барьерами на потенциальной поверхности атомных перегруппировок
внутри углеродных полиэдров.
3/. Движение ионов внутри полиэдров под действием
температурных флюктуаций приводит к размыванию потолка валентной зоны,
состоящей из внедренного электронного состояния и к волне поляризации на
поверхности углеродного полиэдра , которая движется вслед за положительно
заряженными ионами-гостями.
4/. Ненулевой спиновый момент приводит к фиксации атомных
координат ионов-гостей, что согласуется с законом сохранения полного момента
системы. Это свойство должно проявляться как в спектроскопии ЯМР, так
и в спектроскопии ЭПР, влияя на температурную зависимость формы сигналов.
СПИСОК ЛИТЕРАТУРЫ К ГЛАВЕ III.6.
[1] А. В. Елецкий, Б.М. Смирнов,
УФН, 165, 9, 977 (1995).
[2] C.T. White, J.W.
Mintmire, R.C. Mowrwy, D.W. Brenner, D.H. Robertson, J.A. Harrison, B.I.
Dunlop, Predicting properties of fullerenes and their derivatives, in:
Buckminsterfullerenes, edt. By W.E Billups and M.A. Ciufolini, VCH Publishers,
Inc.: NY, 1993.
[3] W. Andreoni, Ann. Rev.
Phys. Chem., 49, 405 (1998).
[4] A.R. Ramirez,
Superconductivity Review, 1, 1&2, 1 (1994)
[5] N. Krawez, A. Gromov, R.
Tellgmann, E.E.B. Campbell, Electronic properties of novel materials - Progress
in molecular nanostructures, XII International Winterschool, Kirchberg, Tyrol,
Austria, 1998, p. 368.
[6] Y. Yoshinari, H. Alloul,
V. Brouet, G. Kriza, K. Holczer, L. Forro, Phys. Rev. B54, 9, 6155 (1996).
[7] T. Pichler, M.S. Golden,
M. Knupfer, J. Fink, XII International Winterschool Electronic properties of
novel materials, Proceedings, edt. by H. Kuzmany, 271 (1998).
[8] D.M. Poirier, M.
Knupfer, J.H. Weaver, W. Andreoni, K. Laasonen, M. Parinello, D.S. Bethune,
K.Kikuchi, Y. Achida, Phys.Rev. B49, 24, 17403 (1994).
[9] J.H. Weaver, Acc. Chem.
Res. 25, 3, 143 (1992).
[10] M.Takata, B.Umeda,
E.Nishibori, M.Sakata, Y.Saito, M.Ohno, H.Shinohara, Nature, 377, 17, 46
(1995).
[11] W. Sato, K. Sueki, K.
Kikuchi, S. Suzuki, Y. Achiba, H. Nakahara, Y. Ohkubo, K. Asai, F. Ambe,
Phys.Rev. B58, 16, 10850 (1998).
[12] C. Gu, F. Stepniak,
D.M. Poirier, M.B. Jost, P.J. Benning,
Y. Chen, T.R. Ohno, J.J.L. Martins, J.H. Weaver, J. Fure, R.E. Smalley, Phys.
Rev. В53,
3, 1196 (1995).
[13] J. Chioslovski, E.D.
Fleischmann, J. Chem. Phys., 94, 5, 3730 (1991).
[14] А.Б. Ройцин, Л.В. Артамонов,
А.А. Климов, ФНТ 23, 10, 1112 (1997).
[15] H. Mauser, T. Clark, A.
Hirsch, XII International Winterschool Electronic properties of novel
materials, Proceedings, edt. by H. Kuzmany, 202 (1998).
[16] C.G. Joslin, J. Yang,
C.G. Gray, J.D. Poll, Chem. Phys. Let., 211, 6, 587 (1993)
[17] L. Pang, F. Brisse, J.
Phys. Chem., 97, 33, 8562 (1993).
[18] Y.S. Li, D. Tomanek,
Chem. Phys. Lett., 221, 5, 453 (1994).
[19] D. Tomanek, Y.S. Li,
Chem. Phys. Lett., 243, 1, 42 (1995).
[20] В.А. Левашов, А.А. Ремова,
В.Р. Белослудов, Письма в ЖЭТФ, 65, 8, 647 (1997).
[21] Y. Maryyama, K. Ohno,
K. Esfarjani, Y. Kawazoe, Sci. Rep. RITU A41, 2, 183 (1996).
[22] A.H.H. Chang, W.C.
Ermler, R.M. Pitzer, J. Chem. Phys., 94, 7, 5004 (1991).
[23] J. Liu, S. Iwata, Phys.
Rev. B50, 8, 5552 (1994).
[24] F. De Proft, C. Van
Alsenoy, P. Geerlings, J. Phys. Chem., 100, 18, 7440 (1996).
[25] S. Patchkovskii, W.
Thiel, J. Chem. Phys., 106, 5, 1796 (1997).
[26] Y. Wang, D. Tomanek,
Chem. Phys. Lett., 208, 1&2, 79 (1993).
[27] B.I. Dunlop, J.L.
Ballester, P.P. Schmidt, J. Phys. Chem., 96, 24, 9781 (1992).
[28]
С.А. Варганов, П.В. Аврамов, С.Г. Овчинников, ФТТ, 42, 2, 378 (2000).
[29] Y. Maniwa, K.
Mizoguchi, K. Kume, K. Kikuchi, I. Ikemoto, S. Suzuki, Y. Achida, Solid_state
Commun., 80, 12, 609 (1991).
[30] R.D. Johnson, C.S.
Yannoni, H.C. Dorn, J.R. Salem, D.S. Bethune, Science, 225, 4, 1235 (1992).
[31] R. Tycko, G. Dabbagh,
R.M. Fleming, R.C. Haddon, A.V. Makhija, S.M. Zahurak, Phys.Rev.Lett., 67, 14,
1886 (1991)
[32] P.A. Heiney, J.E.
Fischer, A.R. McGhi, W.J. Romanov, A.M. Denenstein, J.P. McCauley, Jr., A.B.
Smith III, D.E. Cox, Phys. Rev. Lett., 66, 22, 2911 (1991).
[33] R. Moret, P.A. Albouy,
V. Agafonov, R. Ceolin, D. Andre, A. Dvorkin, H. Szwarc, C. Fabre, A. Rassat,
A. Zahab, P. Bernier, J. Phys. (France) I2, 5, 511 (1992).
[34] W. Andreoni, A.
Curioni, Phys. Rev. Lett., 77, 5, 834 (1996).
[35] W. Andreoni, A.
Curioni, Appl. Phys. A66, 299 (1998).
[36] J.L. Ballester, B.I.
Dunlop, Phys. Rev. A45, 10, 7985 (1992).
[37] M.W. Schmidt, K.K.
Baldridge, J.A. Boatz. J. Comp. Chem. 14, 6, 1347 (1993).